Marfan syndrome is an inherited disorder caused by a mutation in the FBN1 gene that affects the connective tissue, the body’s main support structure. The most prominent symptoms of this condition are disproportionately long limbs in the skeletal system, chest wall deformities, lens displacement in the eyes and, most importantly, aortic aneurysm in the cardiovascular system. Despite its serious life-threatening risks, it can now be successfully managed with effective treatment methods such as drug therapies to protect the aortic vessel and preventive surgery. Early diagnosis and the right treatment approach significantly increase the life expectancy and quality of life of patients.
| Definition | A genetic disorder affecting connective tissue, inherited in an autosomal dominant manner, involving multiple systems |
| Genetic Factor | Mutation in the FBN1 gene (fibrillin-1); disorder associated with TGF-β signaling pathways |
| Affected Systems | Cardiovascular system (aortic root dilatation, aortic dissection), skeletal system (long limbs, scoliosis, pectus deformities), ocular system (lens ectopia), lung (pneumothorax), skin (striae) |
| Symptoms | Tall stature, disproportionately long arms and legs, chest wall deformities, flexible joints, myopia, lens shift, heart murmur, aortic enlargement |
| Diagnostic Methods | Ghent criteria, genetic testing (FBN1 mutation), ECHO, aortic evaluation with MRI/CT, eye examination, skeletal system evaluation |
| Complications | Aortic aneurysm and dissection, mitral valve prolapse, sudden cardiac death, scoliosis, lens dislocation |
| Treatment Methods | Beta blockers or ARB (such as losartan), surgical intervention (e.g. Bentall’s surgery), regular cardiologic and ophthalmologic follow-up |
| Follow-up and Monitoring | Regular measurement of aortic diameter (ECHO/CT/MRI), cardiology and genetic counseling, eye and skeletal system checks |
| Ways of Prevention | Genetic counseling, physical activity limitations (especially heavy sports), regular medical monitoring, prophylactic surgical planning |
What is Marfan Syndrome and how does it affect the body?
To understand Marfan syndrome, you first need to know what connective tissue does. You can think of connective tissue as the steel columns and mortar that make up the skeleton of a building. It is what gives our body its shape, strength and flexibility. It is everywhere, from our bones to our muscles, from our blood vessels to our heart valves. Marfan syndrome is a weakness in this basic building material. For this reason, its effects are not limited to a single organ; wherever there is connective tissue in the body, an effect can be seen. The most noticeable effects occur in structures that are subjected to constant pressure and stress: the heart, large blood vessels (especially the aorta), the skeletal system and the eyes.
Another important feature of this syndrome is that it can vary in severity from person to person. Even within the same family, one sibling may have only mild joint flexibility and myopia, while another sibling may have serious problems requiring emergency heart surgery. This variability best explains why the management of Marfan syndrome needs to be individualized. Although it is rare, it is one of the most common inherited connective tissue disorders, occurring in approximately 3,000-5,000 people.
What is the Genetic Origin of Marfan Syndrome?
Like any genetic disease, Marfan syndrome is caused by a “software error”. This error is in a gene on chromosome 15 called FBN1. The job of this gene is to tell the body how to make a very important protein called fibrillin-1. Fibrillin-1 is the main component of the filamentous networks called microfibrils that make up connective tissue. These networks are like rubber bands that give tissues both strength and elasticity.
A mutation, i.e. an error, in the FBN1 gene leads to either an insufficient amount of this fibrillin protein being produced or its quality being impaired. As a result, connective tissue becomes weak and flimsy. But it is not just a structural weakness. The fibrillin protein also regulates certain signaling molecules (especially TGF-β) that control growth and repair processes in the body. Defective fibrillin causes these signaling molecules to become overactive. This overactivity, in turn, contributes to many of the problems characteristic of Marfan syndrome, such as disproportionate lengthening of bones and further weakening of blood vessel walls. In short, Marfan syndrome is not just a wear and tear problem but an active process caused by abnormal cellular signaling.
How is Marfan Syndrome Transmitted within the Family?
Marfan syndrome is transmitted from generation to generation through an inheritance pattern called “autosomal dominant”. The meaning of this medical term is actually quite simple: The faulty gene only needs to come from one parent for the disease to occur. We can liken it to a coin toss. For each child of a parent with Marfan syndrome, the probability of passing on the gene is P, which is like flipping a coin. Regardless of gender, both boys and girls are equally affected.
In about three quarters (u) of cases, the disease is inherited in this way. In the remaining quarter (%), however, the situation is completely different. Although there is no family history of Marfan syndrome, a new genetic mutation (de novo mutation) may occur spontaneously in the child. This can lead to questions such as “Why did we get this? There was never anything like this in our family”. This is very important information showing that the absence of a family history does not exclude the diagnosis of Marfan syndrome and that the diagnosis should always be based on objective medical findings.
What are the Skeletal Symptoms of Marfan Syndrome?
Perhaps the most easily recognized symptoms of Marfan syndrome are seen in the general structure of the body and the skeletal system. These symptoms may not make sense on their own, but together they raise an important suspicion. The most common skeletal findings are as follows:
- Significantly taller than family
- Disproportionately long arms, legs and fingers
- arachno dactyly, also known as “spider-fingeredness”
- When the arms are spread to the side, the stroke distance exceeds the height
- Inward depression of the breastbone (shoemaker’s chest)
- Protruding breastbone (pigeon breast)
- Sideways curvature of the spine (scoliosis)
- Hunchback (kyphosis)
- Excessive flexibility and mobility in the joints (hypermobility)
- Flat feet (pes planus)
- A long and narrow facial structure
- High and curved palate
- Crowding and crowding of teeth
How Does Marfan Syndrome Affect Eye Health?
After skeletal findings, the most important clues often come from the eyes. The weakness of the connective tissue also affects the delicate structures of the eye. A detailed examination by an ophthalmologist can play a key role in the diagnosis of Marfan syndrome. The main eye findings to pay attention to are as follows:
- Displacement of the lens of the eye (ectopia lentis). This is a very typical finding for Marfan syndrome.
- High degree of myopia (inability to see far away clearly)
- Astigmatism
- Cataract development at an early age
- Increased intraocular pressure (glaucoma)
- Tearing or detachment of the retinal layer (retinal detachment)
Dislocation of the lens of the eye in particular is not just a visual defect; it is also an alarm bell for risks in other parts of the body, especially the heart. It is known that the more pronounced the eye symptoms, the higher the risk of serious problems with the aorta. Therefore, when Marfan is suspected during an eye examination, it is vital that the patient is immediately evaluated by a cardiologist.
What are the Effects of Marfan Syndrome on the Cardiovascular System?
The most serious and life-threatening complications of Marfan syndrome occur in the cardiovascular system. Therefore, the follow-up and treatment of the syndrome is largely managed by a cardiologist and cardiac surgeon. At the heart of the problem is the aorta, the body’s largest artery, which exits the heart. The aorta is the main pipeline that distributes high-pressure blood throughout the body with every beat of the heart. To withstand this enormous pressure, its walls must be very strong and flexible. In Marfan syndrome, the aortic wall loses this resilience due to weakness in the connective tissue. Over time, it gradually begins to expand like a balloon under the influence of the blood pressure inside it. This condition is called “aortic aneurysm”.
The aneurysm itself may initially be asymptomatic, but the real danger is that it grows silently. Once it reaches a certain diameter, this weakened vessel wall can either rupture completely (rupture) or tear through its inner layers, causing blood to leak between the vessel walls (aortic dissection). Both conditions require urgent surgical intervention and can be fatal. Therefore, the main goal of Marfan syndrome follow-up is to detect this “time bomb” before it explodes and to defuse it with preventive surgery.
What are the Other Health Problems in Marfan Syndrome?
Although the heart, skeleton and eyes are the most commonly affected systems, Marfan syndrome can also cause problems in other areas where connective tissue is present. These symptoms form other important pieces of the diagnostic puzzle.
- Lungs There is an increased risk of sudden lung collapse (spontaneous pneumothorax) due to weakness of the air sacs in the lung.
- Skin: Stretch marks (striae) may occur, especially on the shoulders, back and buttocks, without a clear history of weight gain or loss.
- Nervous System: Enlargement of the membrane surrounding the spinal cord, called dura mater, in the lumbar region (dural ectasia) is an important finding that can cause chronic low back, back and leg pain.
According to Which Criteria is Marfan Syndrome Diagnosed?
Marfan syndrome cannot be diagnosed based on a single blood test or imaging study. It is a detailed detective work that requires doctors from different specialties to come together. The diagnostic process uses an international guideline known as the “Revised Ghent Criteria”, updated in 2010. This modern approach builds the diagnosis on the two most critical findings: Aortic root enlargement and displacement of the lens of the eye (ectopia lentis).
The pathways to diagnosis are usually as follows:
- In a person without a family history of Marfan, the diagnosis is certain if both aortic dilatation and a shift in the lens of the eye are present.
- If aortic dilatation is detected and FBN1 mutation is found in genetic testing, the diagnosis is definitive.
- If only aortic dilatation is present, the diagnosis requires an evaluation of findings in other body systems according to a scoring system. If 7 or more points are accumulated in this so-called “systemic score”, a diagnosis is made.
This systemic scoring shows how many small findings that at first glance may seem unrelated (flat feet, skin cracks, facial features, etc.) can be significant when taken together, proving why the diagnosis requires a holistic approach.
What is the Role of Drugs and Lifestyle in the Treatment of Marfan Syndrome?
There is not yet a treatment that completely eliminates Marfan syndrome. However, modern medicine offers very effective methods to slow the progression of the disease and prevent the most dangerous complication, aortic rupture. Two main groups of drugs form the basis of treatment. These medicines aim to slow the rate at which the aneurysm grows by reducing stress on the aortic wall.
- Beta-blockers: They lower blood pressure by reducing the heart rate and the force of contraction of the heart.
- Angiotensin Receptor Blockers (ARBs): They lower blood pressure by relaxing blood vessels and are also thought to provide additional protection to connective tissue via the TGF-β signaling pathway.
Another issue that is as important as medication is lifestyle adjustments. The aim here is to avoid activities that can cause sudden and severe increases in blood pressure.
Recommended activities to avoid:
- Lifting heavy weights
- Competitive and contact sports (soccer, basketball, combat sports)
- Isometric exercises that require intense effort (such as push-ups, sit-ups)
Activities that are generally considered safe:
- Brisk walking
- Swimming
- Cycling (at a non-strenuous pace)
- Golf
- Bowling
The most appropriate activity plan for each patient should be determined individually by the cardiologist following the patient.
When and Why Is Preventive Heart Surgery Necessary in Marfan Syndrome?
Despite medication and lifestyle changes, the enlargement of the aorta may continue. When the aortic diameter reaches a critical threshold at which the risk of rupture becomes unacceptably high, “preventive” or prophylactic surgery is considered. The philosophy here is not to wait for the disaster to happen, but to eliminate the risk by acting before it does.
The most important criterion for the decision to operate is the diameter of the aortic root. The generally accepted thresholds are:
- When the aortic diameter reaches 5.0 cm in patients without risk factors
- At 4.5 cm if there are additional risk factors such as family history of aortic dissection, rapid growth, severe valvular insufficiency
- In women planning pregnancy, in the range of 4.0-4.5 cm according to the risk status
These figures are a guide and the final decision is made jointly by the patient and the surgeon, taking into account the patient’s general condition, body measurements and other individual factors.
What are the Cardiac Surgery Methods for Marfan Syndrome?
There are two main surgical approaches currently used to replace the aortic root. The choice depends on the condition of the patient’s aortic valve and the surgeon’s experience.
- Bentall Procedure (Composite Graft Exchange): This is a traditional and very effective procedure. In the operation, the enlarged aortic root and the patient’s own aortic valve, which is often damaged, are removed together. It is replaced with a composite structure consisting of a synthetic tube (graft) and a prosthetic valve (mechanical or biological) sewn onto it. The main advantage of this method is its durability, whereas the disadvantage is that the prosthetic valve means that the patient will either have to take blood thinners for life (mechanical valve) or undergo another operation in the future (biological valve).
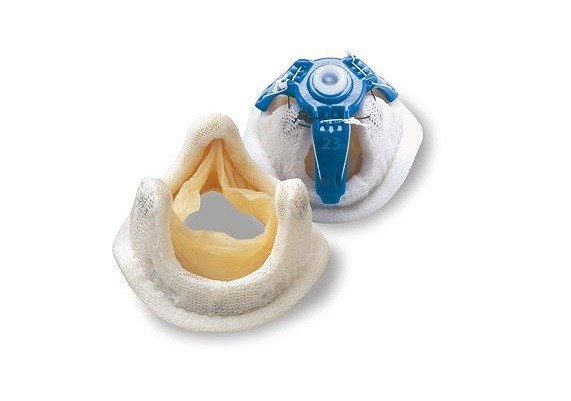
- Valve-sparing Aortic Root Replacement (David Procedure): This is a more modern and technically more challenging approach. If the patient’s own aortic valve is intact, only the enlarged aortic wall is replaced with a synthetic graft, while the patient’s own healthy valve is preserved and reinserted into the new graft. This is a “restoration” procedure rather than a repair. Its biggest advantage is that it saves the patient from all the difficulties of prosthetic valves and the use of blood thinners. It makes a tremendous difference in terms of quality of life, especially for young patients.
What is the Life Expectancy of a Person with Marfan Syndrome?
This is one of the questions that patients and their families are most curious and concerned about: “How many years does Marfan syndrome live?” The answer to this question reveals the incredible progress that modern medicine has made in the last 50 years. in the 1970s, the average life expectancy of a person with Marfan syndrome was unfortunately limited to the 30s or 40s. Today, the picture is completely different. Thanks to accurate diagnosis, regular follow-up, effective drug therapies and, most importantly, timely preventive aortic surgery, the life expectancy of individuals with Marfan syndrome has increased to near-normal levels, i.e. to the 70s and above.
Here are the key steps that made this historic achievement possible:
- Increase in early and accurate diagnosis rates
- Widespread use of aortic-protecting drug therapies
- Close follow-up with advances in imaging methods
- Development of safe and effective preventive aortic surgery techniques
Now that patients are living longer, other effects of Marfan syndrome (such as weakness of the heart muscle, aneurysms in other parts of the aorta, chronic joint problems) are being encountered in later life. This shows once again that Marfan management is indeed a lifelong, dynamic process and that specialist follow-up should never be abandoned. In this journey, psychological support and a holistic approach centering on the patient’s quality of life are as important as medical treatments.
Frequently Asked Questions
What is Marfan syndrome?
Marfan syndrome is a genetic disorder that affects connective tissue. It can affect the heart, blood vessels, skeletal system, eyes and lungs.
Which gene is affected?
A mutation in the FBN1 (fibrillin-1) gene causes this syndrome.
How does it affect the heart and blood vessels?
Aortic dilatation can lead to serious cardiovascular complications such as aortic aneurysm and mitral valve prolapse.
Is Marfan syndrome hereditary?
Yes, it is inherited as an autosomal dominant trait; it can be inherited from the mother or father, or it can be caused by a new mutation.
What are his physical characteristics?
Typical findings include tall stature, long fingers (arachnodactyly), collapsed or protruding sternum, scoliosis and thin build.
Are eye problems visible?
Yes, problems such as lens shift (ectopia lentis), myopia and retinal detachment are common.
How is it diagnosed?
The diagnosis is confirmed by physical examination, genetic testing, ECHO, eye and skeletal examination.
Is there a cure for Marfan syndrome?
There is no cure; a multidisciplinary approach is needed to manage symptoms.
Which complications are life-threatening?
Aortic dissection (rupture) is the most serious and fatal complication and requires immediate intervention.
Which specialists are involved in the treatment process?
Cardiologists, orthopedists, ophthalmologists and geneticists work in collaboration.
Can people with Marfan syndrome play sports?
Heavy sports should be avoided; low-risk exercises (swimming, walking) should be preferred.
Is pregnancy risky?
Yes, the risk is increased, especially if there is aortic dilatation; cardiology and obstetrician-gynecologist control is essential.
Can people with Marfan syndrome lead normal lives?
Regular follow-up and appropriate treatment can improve quality of life and prolong life.
Can this syndrome be recognized in children?
Yes, it can be diagnosed in childhood if there are physical symptoms and a family history.
Is surgical treatment necessary in Marfan syndrome?
Aortic surgery is recommended if aortic dilatation reaches a critical size.

Prof. Dr. Yavuz Beşoğul graduated from Erciyes University Faculty of Medicine in 1989 and completed his specialization in Cardiovascular Surgery in 1996. Between 1997 and 2012, he served at Eskişehir Osmangazi University Faculty of Medicine as Assistant Professor, Associate Professor, and Professor, respectively. Prof. Dr. Beşoğul, one of the pioneers of minimally invasive cardiovascular surgery in Türkiye, has specialized in closed-heart surgeries, underarm heart valve surgery, beating-heart bypass, and peripheral vascular surgery. He worked at Florence Nightingale Kızıltoprak Hospital between 2012–2014, Medicana Çamlıca Hospital between 2014–2017, and İstinye University (Medical Park) Hospital between 2017–2023. With over 100 publications and one book chapter, Prof. Dr. Beşoğul has contributed significantly to the medical literature and is known for his minimally invasive approaches that prioritize patient safety and rapid recovery.